To study DNA or RNA, there are a number of “wet-lab” (laboratory) and “dry-lab” (analysis) steps which are required to access the genetic code from inside cells, polish it to a high-sheen such that the delicate technology we rely on can use it, and then make sense of it all. Destructive enzymes must be removed, one strand of DNA must be turned into millions of strands so that collectively they create a measurable signal for sequencing, and contamination must be removed. Yet, what constitutes contamination, and when or how to deal with it, remains an actively debated topic in science. Major contamination sources include human handlers, non-sterile laboratory materials, other samples during processing, and artificial generation due to technological quirks.
Contamination from human handlers
This one is easiest to understand; we constantly shed microorganisms and our own cells and these aerosolized cells may fall into samples during collection or processing. This might be of minimal concern working with feces, where the sheer number of microbial cells in a single teaspoon swamp the number that you might have shed into it, or it may be of vital concern when investigating house dust which not only has comparatively few cells and little diversity, but is also expected to have a large amount of human-associated microorganisms present. To combat this, researchers wear personal protective equipment (PPE) which protects you from your samples and your samples from you, and work in biosafety cabinets which use laminar air flow to prevent your microbial cloud from floating onto your workstation and samples.

Image Credit: Kristina Drobny
Fun fact, many photos in laboratories are staged, including this one, of me as a grad student. I’m just pretending to work. Reflective surfaces, lighting, cramped spaces, busy scenes, and difficulty in positioning oneself makes “action shots” difficult. That’s why many lab photos are staged, and often lack PPE.
Contamination from laboratory materials
Microbiology or molecular biology laboratory materials are sterilized before and between uses, perhaps using chemicals (ex. 70% ethanol), an ultraviolet lamp, or autoclaving which combines heat and pressure to destroy, and which can be used to sterilize liquids, biological material, clothing, metal, some plastics, etc. However, microorganisms can be tough – really tough, and can sometimes survive the harsh cleaning protocols we use. Or, their DNA can survive, and get picked up by sequencing techniques that don’t discriminate between live and dead cellular DNA.
In addition to careful adherence to protocols, some of this biologically-sourced contamination can be handled in analysis. A survey of human cell RNA sequence libraries found widespread contamination by bacterial RNA, which was attributed to environmental contamination. The paper includes an interesting discussion on how to correct this bioinformatically, as well as a perspective on contamination. Likewise, you can simply remove sequences belonging to certain taxa during quality control steps in sequence processing. There are a number of hardy bacteria that have been commonly found in laboratory reagents and are considered contaminants, the trouble is that many of these are also found in the environment, and in certain cases may be real community members. Should one throw the Bradyrhizobium out with the laboratory water bath?

Chimeras
Like the mythical creatures these are named for, sequence chimeras are DNA (or cDNA) strands which are accidentally created when two other DNA strands merged. Chimeric sequences can be made up of more than two DNA strand parents, but the probability of that is much lower. Chimeras occur during PCR, which takes one strand of genetic code and makes thousands to millions of copies, and a process used in nearly all sequencing workflows at some point. If there is an uneven voltage supplied to the machine, the amplification process can hiccup, producing partial DNA strands which can concatenate and produce a new strand, which might be confused for a new species. These can be removed during analysis by comparing the first and second half of each of your sequences to a reference database of sequences. If each half matches to a different “parent”, it is deemed chimeric and removed.
Cross-sample contamination
During DNA or RNA extraction, genetic code can be flicked from one sample to another during any number of wash or shaking steps, or if droplets are flicked from fast moving pipettes. This can be mitigated by properly sealing all sample containers or plates, moving slowly and carefully controlling your technique, or using precision robots which have been programmed with exacting detail — down to the curvature of the tube used, the amount and viscosity of the liquid, and how fast you want to pipette to move, so that the computer can calculate the pressure needed to perform each task. Sequencing machines are extremely expensive, and many labs are moving towards shared facilities or third-party service providers, both of which may use proprietary protocols. This makes it more difficult to track possible contamination, as was the case in a recent study using RNA; the researchers found that much of the sample-sample contamination occurred at the facility or in shipping, and that this negatively affected their ability to properly analyze trends in the data.
Sample-sample contamination during sequencing
Controlling sample-sample contamination during sequencing, however, is much more difficult to control. Each sequencing technology was designed with a different research goal in mind, for example, some generate an immense amount of short reads to get high resolution on specific areas, while others aim to get the longest continuous piece of DNA sequenced as possible before the reaction fails or become unreliable. they each come with their own quirks and potential for quality control failures.
Due to the high cost of sequencing, and the practicality that most microbiome studies don’t require more than 10,000 reads per sample, it is very common to pool samples during a run. During wet-lab processing to prepare your biological samples into a “sequencing library”, a unique piece of artificial “DNA” called a barcode, tag, or index, is added to all the pieces of genetic code in a single sample (in reality, this is not DNA but a single strand of nucleotides without any of DNA’s bells and whistles). Each of your samples gets a different barcode, and then all your samples can be mixed together in a “pool”. After sequencing the pool, your computer program can sort the sequences back into their respective samples using those barcodes.
While this technique has made sequencing significantly cheaper, it adds other complications. For example, Illumina MiSeq machines generate a certain number of sequence reads (about 200 million right now) which are divided up among the samples in that run (like a pie). The samples are added to a sequencing plate or flow cell (for things like Illumina MiSeq). The flow cells have multiple lanes where samples can be added; if you add a smaller number of samples to each lane, the machine will generate more sequences per sample, and if you add a larger number of samples, each one has fewer sequences at the end of the run. you have contamination. One drawback to this is that positive controls always sequence really well, much better than your low-biomass biological samples, which can mean that your samples do not generate many sequences during a run or means that tag switching is encouraged from your high-biomass samples to your low-biomass samples.
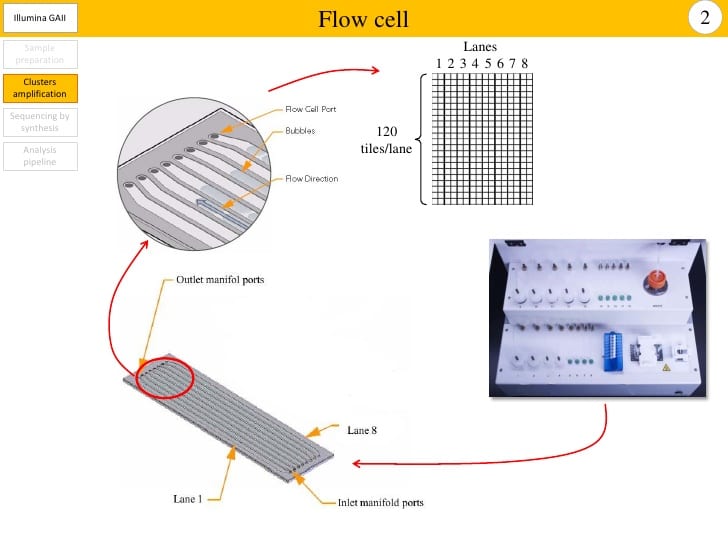

Cross-contamination can happen on a flow cell when the sample pool wasn’t thoroughly cleaned of adapters or primers, and there are great explanations of this here and here. To generate many copies of genetic code from a single strand, you mimic DNA replication in the lab by providing all the basic ingredients (process described here). To do that, you need to add a primer (just like with painting) which can attach to your sample DNA at a specific site and act as scaffolding for your enzyme to attach to the sample DNA and start adding bases to form a complimentary strand. Adapters are just primers with barcodes and the sequencing primer already attached. Primers and adapters are small strands, roughly 10 to 50 nucleotides long, and are much shorter than your DNA of interest, which is generally 100 to 1000 nucleotides long. There are a number of methods to remove them, but if they hang around and make it to the sequencing run, they can be incorporated incorrectly and make it seem like a sequence belongs to a different sample.
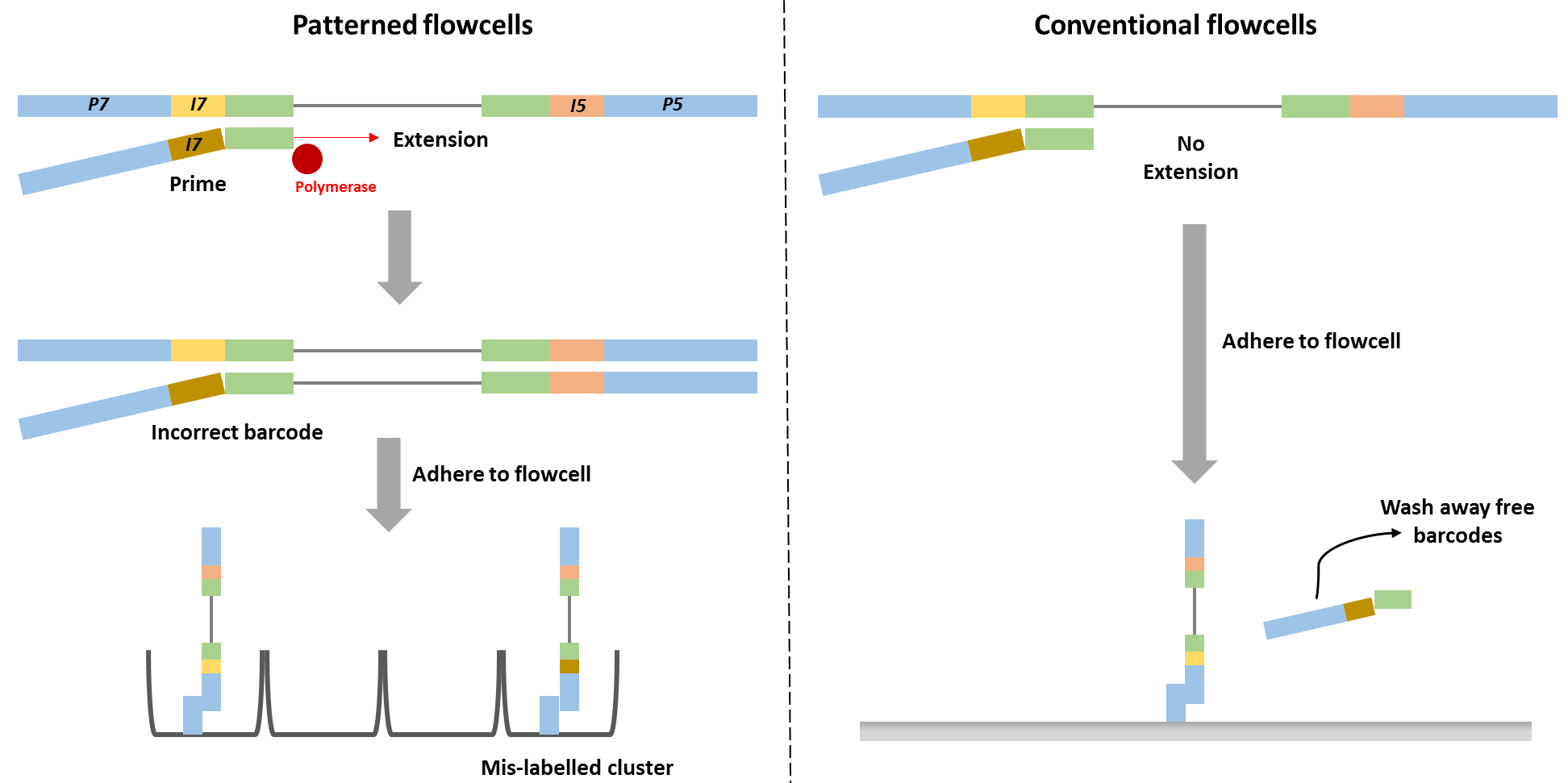
This may sound easy to fix, but sequencing library preparation already goes through a lot of stringent cleaning procedures to remove everything but the DNA (or RNA) strands you want to work with. It’s so stringent, that the problem of barcode swapping, also known as tag switching or index hopping, was not immediately apparent. Even when it is noted, it typically affects a small number of the total sequences. This may not be an issue, if you are working with rumen samples and are only interested in sequences which represent >1% of your total abundance. But it can really be an issue in low biomass samples, such as air or dust, particularly in hospitals or clean rooms. If you were trying to determine whether healthy adults were carrying but not infected by the pathogen C. difficile in their GI tract, you would be very interested in the presence of even one C. difficile sequence and would want to be extremely sure of which sample it came from. Tag switching can be made worse by combining samples from very different sample types or genetic code targets on the same run.
There are a number of articles proposing methods of dealing with tag switching using double tags to reduce confusion or other primer design techniques, computational correction or variance stabilization of the sequence data, identification and removal of contaminant sequences, or utilizing synthetic mock controls. Mock controls are microbial communities which have been created in the lab by mixed a few dozen microbial cultures together, and are used as a positive control to ensure your procedures are working. because you are adding the cells to the sample yourself, you can control the relative concentrations of each species which can act as a standard to estimate the number of cells that might be in your biological samples. Synthetic mock controls don’t use real organisms, they instead use synthetically created DNA to act as artificial “organisms”. If you find these in a biological sample, you know you have contamination. One drawback to this is that positive controls always sequence really well, much better than your low-biomass biological samples, which can mean that your samples do not generate many sequences during a run or means that tag switching is encouraged from your high-biomass samples to your low-biomass samples.
Incorrect base calls
Cross-contamination during sequencing can also be a solely bioinformatic problem – since many of the barcodes are only a few nucleotides (10 or 12 being the most commonly used), if the computer misinterprets the bases it thinks was just added, it can interpret the barcode as being a different one and attribute that sequence to being from a different sample than it was. This may not be a problem if there aren’t many incorrect sequences generated and it falls below the threshold of what is “important because it is abundant”, but again, it can be a problem if you are looking for the presence of perhaps just a few hundred cells.
Implications
When researching environments that have very low biomass, such as air, dust, and hospital or cleanroom surfaces, there are very few microbial cells to begin with. Adding even a few dozen or several hundred cells can make a dramatic impact into what that microbial community looks like, and can confound findings.
Collectively, contamination issues can lead to batch effects, where all the samples that were processed together have similar contamination. This can be confused with an actual treatment effect if you aren’t careful in how you process your samples. For example, if all your samples from timepoint 1 were extracted, amplified, and sequenced together, and all your samples from timepoint 2 were extracted, amplified, and sequenced together later, you might find that timepoint 1 and 2 have significantly different bacterial communities. If this was because a large number of low-abundance species were responsible for that change, you wouldn’t really know if that was because the community had changed subtly or if it was because of the collective effect of low-level contamination.
Stay tuned for a piece on batch effects in sequencing!